Autor/a: William F. Young, Jr The Incidentally Discovered Adrenal Mass. N Engl J Med 2007;356:601-10
Caso clínico
En una tomografía computarizada (TC) del abdomen, realizada a una mujer de 68 años para evaluar un malestar en el hipocondrio derecho aún sin diagnóstico, se halló incidentalmente un tumor de 2,8 cm de diámetro en la glándula suprarrenal izquierda. Su historia clínica solo muestra hipertensión arterial, la cual está bien controlada con 25 mg diarios de hidroclorotiazida. No relata sudoración, palpitaciones, cefaleas, aumento de peso o debilidad de los músculos proximales. ¿Cuál es la evaluación que debe hacerse?
El problema clínico
Un “incidentaloma” suprarrenal (IS) es un tumor suprarrenal, generalmente de 1cm o más de diámetro, que es descubierto en forma accidental durante un examen radiológico realizado para el estudio de otras patologías diferentes a la enfermedad suprarrenal. Esta definición excluye los casos en los se ha “perdido” el diagnóstico de un síndrome adrenal-dependiente sintomático debido a una entrevista o un examen físico superficiales. El uso tan difundido de la ecografía abdominal, la TC no contrastada y las imágenes por resonancia magnética (IRM) ha creado el dilema clínico del IS. Muchos estudios en autopsias han examinado la frecuencia de los nódulos de IS, llegando al 6% en un trabajo que analizó 87.065 autopsias. Recientemente se comprobó que las TC abdominales dan resultados parecidos (4%). La prevalencia de los adenomas suprarrenales aumenta con la edad: es más raro en jóvenes (0,2% en personas de 20 a 29 años) y llega hasta un 7% en personas mayores de 70 años.
Clínicamente, la mayoría de los IS son adenomas suprarrenales benignos no hipersecretores. Otro diagnóstico frecuente es el adenoma suprarrenal corticosecretor, el feocromocitoma, el carcinoma suprarrenal y el carcinoma metastásico.
Estrategias y evidencias
Todavía no se ha establecido cuál es mejor abordaje diagnóstico para un paciente con IS. Sin embargo, dice el autor, es razonable comenzar con un interrogatorio y un examen físico minuciosos, enfocados en los signos y síntomas sugestivos de hiperfunción suprarrenal o enfermedad maligna y, análisis hormonales.
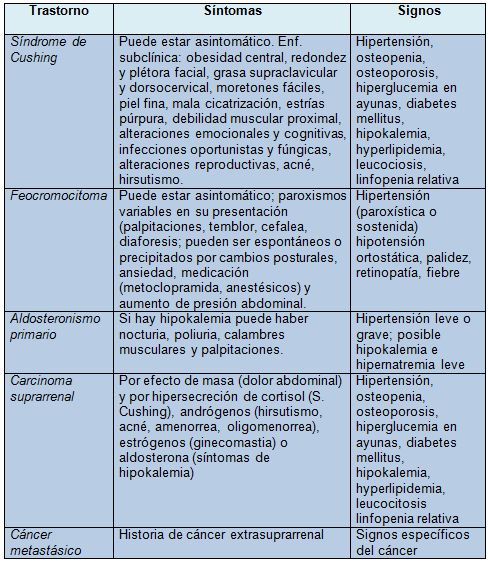
Síntomas y signos sugestivos de hiperfunción suprarrenal o enfermedad maligna
Aunque no se ha hecho la validación prospectiva de un enfoque diagnóstico específico, el autor presenta un algoritmo basado en la experiencia clínica, con datos de laboratorio y estudios por imágenes.
Evaluación hormonal
Síndorme de Cushing subclínico
El término síndrome de Cushing subclínico se refiere a la secreción autónoma de cortisol (independiente del control hipotálamo-hipofisario normal) en pacientes sin signos y síntomas típicos de hipercortisolismo. Aunque los estigmas obvios del síndrome están ausentes, estos pacientes puede sufrir los efectos adversos de la secreción endógena continua de cortisol, como hipertensión arterial, obesidad, diabetes mellitus y osteoporosis.
En un informe que resume los resultados de 13 estudios con 2005 pacientes con IS, el 5,3% de ellos presentaba secreción autónoma de cortisol. Como estos pacientes no presentaban un síndrome de Cushing clínico y la excreción de cortisol urinario era normal, se justifica hacer su determinación al comenzar el estudio del paciente. Debido a que con la mayoría de los análisis con corticotrofina disponibles en el comercio no hay manera de distinguir entre los valores bajos y normales y los valores suprimidos, la autonomía suprarrenal se evalúa mejor mediante la prueba de supresión nocturna con 1 mg de dexametasona.
Aunque el valor de corte óptimo sigue siendo un tema de debate, según esta prueba se consideran anormales los valores de cortisol superiores a los 5 µg/dL (138 mmol/L); la especificidad de la prueba es del 91%; si el resultado es anormal, debe ser confirmado para descartar los positivos falsos. Los datos de los trabajos aleatorizados carecen de una guía para el manejo óptimo del síndrome de Cushing subclínico. El autor sostiene que una estrategia conveniente es considerar la adrenalectomía en los pacientes más jóvenes (menores de 40 años) y en aquellos con trastornos potencialmente atribuibles a la secreción autónoma de glucocorticoides (por ej., el comienzo o el empeoramiento reciente de la hipertensión, la diabetes mellitus, la obesidad o la osteoporosis). Un paciente con síndrome de Cushing subclínico debe recibir tratamiento glucocorticoide perioperatorio debido al riesgo de insuficiencia suprarrenal, crisis hemodinámica o muerte. En el posoperatorio, se debe evaluar la necesidad de un reemplazo hormonal prolongado y el descenso escalonado de los glucocorticoides exógenos. En pocas series de casos, luego de la adrenalectomía unilateral en pacientes con síndrome de Cushing subclínico se reportó pérdida de peso, mejoría de la hipertensión, control glucémico, o ambos, y la normalización de los marcadores del intercambio óseo.
Se requieren estudios prospectivos a largo plazo para tener una mayor comprensión de la historia natural de este síndrome y mejores normas para la toma de decisiones, en vista a una intervención quirúrgica. Dos estudios han sugerido que la secreción de cortisol en el momento en que se descubre el IS puede ser normal pero que se puede tornar autónoma en los 4 o más años siguientes. Por eso, es conveniente repetir el screening hormonal anualmente, durante 4 años, como lo aconseja el National Institutes of Health (NIH) .
Feocromocitoma clínicamente silente
Aproximadamente el 5% de los IS son feocromocitomas. En un estudio, 19 de 33 feocromocitomas suprarrenales (58%) fueron detectados como IS, y solo 10 de 19 pacientes tenían hipertensión. Sin embargo, aun los feocromocitomas silentes pueden ser letales.
Las características de un tumor suprarrenal en las imágenes puede ser útil para determinar si es un feocromocitoma aunque ellas no hacen diagnóstico de feocromocitoma. Ellas son: mayor atenuación en la tomografía computarizada sin contraste, hipervascularización importante del tumor, lavado tardío del medio de contraste, señal de intensidad elevada en las imágenes por resonancia magnética (IRM) de T2.
Debido a que no todos los feocromocitomas tienen su fenotipo y a que la experiencia de los radiólogos y clínicos para su identificación puede variar, es necesario recurrir a la evaluación bioquímica en todos los pacientes. Se recomienda la determinación de metanefrinas fraccionadas y catecolaminas en una muestra de orina de 24 horas. Los valores elevados de alguna de ellas o de ambas es altamente sensible y específico de feocromocitoma (91 a 98% en series de la Clínica Mayo). La determinación adicional de catecolaminas fraccionadas en orina de 24 horas es muy útil para el diagnóstico en pacientes con neoplasias secretoras de dopamina. Ante la sospecha elevada de feocromocitoma subclínico basada en el fenotipo de las imágenes pero con estudios urinarios normales, es útil la determinación de metanefrinas plasmáticas libres fraccionadas. Este análisis tienen sensibilidad elevada (96 a 100%) pero especificidad baja (85 a 89%; 77% en mayores de 60 años). Por lo tanto, esta prueba solo se justifica cuando la sospecha de feocromocitoma es elevada, para minimizar el riesgo de resultados positivos falsos que podrían llevar a una cirugía innecesaria.
Aldosteronismo primario
Aproximadamente el 1% de los IS son adenomas productores de aldosterona. La secreción excesiva de aldosterona se asocia con mayor riesgo de enfermedad cardiovascular y otros trastornos; en estos pacientes, es necesario normalizar los niveles de aldosterona circulante o bloquear los receptores de mineralocorticoides. A todos los pacientes con IS hipertensos se les debe investigar el hiperaldosteronismo. Dado que los pacientes con adenomas productores de aldosterona pueden tener una potasemia normal, su determinación no es útil para el screening. Más adecuado es establecer la relación entre la concentración matinal de aldosterona y la actividad de la renina, ambas en plasma. Si la relación es elevada se debe confirmar el diagnóstico de aldosteronismo primario mediante la investigación de la secreción autónoma de mineralocorticoides.
Otros procesos con actividad hormonal
Los tumores suprarrenales secretores de hormonas sexuales son raros y casi siempre tienen manifestaciones clínicas (hirsutismo o virilización). Por lo tanto, no es necesario hacer el estudio sistemático para el exceso de andrógenos o estrógenos en pacientes con IS.
La hiperplasia suprarrenal congénita no clásica (uni o bilateral) es otra causa infrecuente de IS. No se recomienda la realización sistemática de las pruebas de estimulación de cosintrofina con la medición de los precursores del cortisol (17-hidroxiprogesterona) sino que deben reservarse para los pacientes cuyo diagnóstico se sospecha por la presencia de manifestaciones clínicas (hiperandrogenismo) o de tumores suprarrenales bilaterales.
Evaluación del potencial maligno
La posibilidad de una enfermedad maligna es el problema mayor que plantea el hallazgo de un IS. Entre los 2005 pacientes con diagnóstico de IS mencionados, en el 4,7% se comprobó un carcinoma suprarrenal y de cáncer metastásico en el 2,5%. El tamaño del tumor y su aspecto en las imágenes son dos predictores importantes de enfermedad maligna.
Tamaño del tumor suprarrenal
Un informe sobre 887 pacientes con IS sostiene que los tumores mayores de 4 cm tienen un 90% de sensibilidad para diagnosticar el carcinoma suprarrenal, pero baja especificidad; solo el 24% de las lesiones mayores de 4 cm fueron malignas. El tamaño también es importante porque cuanto más pequeños son los carcinomas suprarrenales en el momento del diagnóstico, más bajo es el estadio tumoral y mejor es el pronóstico. Aunque la mayoría de los especialistas recomendaría la resección de los tumores suprarrenales mayores de 6 cm de diámetro, las decisiones sobre tratamiento quirúrgico debe también tener en cuenta el fenotipo en las imágenes como así la edad del paciente y cualquier enfermedad coexistente. Por ejemplo, un IS no funcionante mayor de 6,5 cm de diámetro y aspecto radiológico benigno puede ser seguido bajo observación en un octogenario.
Debido a que la prevalencia de los adenomas suprarrenales benignos aumenta con la edad, el hallazgo de un tumor adrenal no funcionantes de 3,2 cm de diámetro en un paciente más joven (menor de 30 año) debe aumentar la sospecha de otro diagnóstico. El tamaño de un IS no afecta las recomendaciones de las pruebas bioquímicas.
Fenotipo en las imágenes
Los signos de la TC no contrastada utilizados para diferenciar los adenomas de los no adenomas dependen del contenido lipídico del tumor y de la rapidez con que desaparece el medio de contraste.
En los adenomas, la grasa intracitoplasmática produce una atenuación baja en la TC no contrastada mientras que los no adenomas producen una atenuación más elevada. En la TC contrastada retardada, los adenomas muestran un lavado rápido del medio de contraste, mientras que los tumores suprarrenales no adenomas tienen un lavado retardado. Diez minutos después de la administración del medio de contraste, se lava más del 50% del medio de contraste, lo que da una sensibilidad y especificidad del 100% para el diagnóstico de adenoma, como se comprobó comparando pacientes con adenomas y aquellos con carcinomas, feocromocitomas o metástasis. Aunque el fenotipo de la imagen no es predictivo de la función hormonal, predice la patología subyacente: los pacientes con imágenes de IS sospechosas son candidatos para la resección quirúrgica.
Enfermedad metastásica
En la mitad de los pacientes con IS que tienen una historia de enfermedad maligna, la causa de los IS es la enfermedad metastásica. Los tumores que con más frecuencia producen metástasis suprarrenales (por lo general, bilaterales) son los de pulmón, riñón, colon, mama, esófago, páncreas, hígado y estómago. En estos casos, cuando se descubre el IS, ya está hecho el diagnóstico del cáncer primitivo; es muy raro descubrir primero el incidentaloma.
La tomografía por emisión de positrones (PET) con 18 fluorodesoxiglucosa (18F-FDG) puede ser útil en pacientes seleccionados porque tiene una sensibilidad elevada para detectar enfermedades malignas. Sin embargo, el 16% de las lesiones suprarrenales benignas puede tener más captación del contraste. La ausencia de actividad en la PET contrastada con 11C-metomidato es específica para los tumores de origen no suprarrenales (feocromocitomas y enfermedad metastásica) pero estas técnicas no son de uso común. Debido a su costo y la falta de datos suficientes para su aplicación sistemática, ambos tipos de PET no están recomendados para la evaluación de los pacientes con IS pero historia de enfermedad maligna.
Biopsia por aspiración con aguja fina
El papel principal de esta biopsia es diferenciar entre el tejido suprarrenal y los tejidos no suprarrenales (metástasis o infección). La biopsia por aspiración con aguja fina (BAAF) guiada por imágenes es relativamente segura, siendo la tasa de complicaciones de 2,8% en una serie de 277 biopsias. Los riesgos de este procedimiento son: el hematoma suprarrenal, el dolor abdominal, la pancreatitis, el neumotórax, la formación de un absceso suprarrenal y la recurrencia del tumor a lo largo del trayecto de la aguja. En un feocromocitoma, la BAAF puede provocar hemorragia y crisis hipertensiva, por lo que hay que tener en cuenta que siempre existe la posibilidad de descartar el feocromocitoma por análisis clínicos antes de hacer la biopsia.
Tumores suprarrenales bilaterales
Cuando los tumores suprarrenales son bilaterales (15% de los IS), los diagnósticos más probables son la enfermedad maligna, la hiperplasia suprarrenal congénita, los adenomas corticales bilaterales y la enfermedad infiltrante de las glándulas suprarrenales. Los pacientes con tumores suprarrenales bilaterales pueden presentar hipofunción suprarrenal, de manera que en tales pacientes es conveniente investigar dicha hipofunción.
Áreas de incertidumbre
La frecuencia y la duración más indicadas para el seguimiento de los pacientes con IS no están establecidas y los datos prospectivos que avalen una guía clínica son escasos. Se recomienda repetir las imágenes a los 6, 12 y 24 meses, pero cuando el aspecto radiológico es sospechoso se aconseja hacerlo a los 3 meses del seguimiento. Por otra parte, tampoco se conoce la relación costo eficacia de la repetición de las imágenes. “Según nuestra experiencia sobre 9 pacientes no publicados,” dice el autor, “la velocidad típica de crecimiento del feocromocitoma suprarrenal benigno es de aproximadamente 0,5 a 1 cm de diámetro por año, mientras que los carcinomas suprarrenales tienen un crecimiento más rápido (>2cm por año).” Sin embargo, acota, la mayoría de los tumores suprarrenales que crece no son malignos.
Durante el seguimiento, puede observarse la aparición de una función suprarrenal anormal (secreción de glucocorticoides y catecolaminas) que no estaba presente al comienzo del estudio, lo que justifica la repetición de los análisis anualmente durante 4 años. Tampoco se conoce la relación costo eficacia de esta conducta.
Guías
Las sociedades profesionales no han publicado guías completas para la evaluación de los pacientes con IS. Las recomendaciones que brinda el autor concuerdan con las publicadas por el NIH en 2003.
Conclusiones y recomendaciones
Para el caso de la paciente relatado al principio, la historia y el examen físico permitirán evaluar la evidencia de un exceso hormonal. Se podría hacer una prueba de supresión nocturna con 1 mg de dexametasona, con recolección de orina de 24 horas para la determinación de metanefrinas fraccionadas y catecolaminas y (debido a que la paciente es hipertensa) también de la concentración de aldosterona y la actividad de la renina plasmáticas. Si los resultados del análisis hormonal inicial corresponden a la secreción autónoma hormonal, y si este hallazgo es confirmado por otros estudios, se debe considerar la realización de una adrenalectomía laparoscópica unilateral. Las imágenes suprarrenales deben ser analizadas por un radiólogo. Si el fenotipo del tumor hace sospechar una infección o enfermedad metastásica, se hará una BAAF (después de descartar el feocromocitoma por pruebas bioquímicas). Si los resultados hormonales son normales y los signos radiológicos son de benignidad, se recomendaría repetir las imágenes a los 6, 12 y 24 meses, y los análisis hormonales 1 vez por año durante 4 años.
Aunque también faltan datos que indiquen si la cirugía es necesaria, se podría considerar la adrenalectomía en el caso que el tumor suprarrenal tenga 4 cm o más, o si se agranda a razón de 1 cm o más durante el período de observación o, hay evidencia de desarrollo de secreción hormonal anormal.
Algoritmo de evaluación para pacientes con incidentaloma suprarrenal
El algoritmo debe aplicarse acorde con la clínica, el aspecto del tumor en las imágenes, la edad del paciente y sus preferencias. Dada la asociación estrecha entre los signos radiológicos y el feocromocitoma, en los pacientes cuyas imágenes hacen sospechar un feocromocitoma (aún con pruebas bioquímicas normales), algunos aconsejan hacer el bloqueo adrenérgico y la resección del tumor.

♦ Comentario y resumen objetivo: Dra. Marta Papponetti