Rev Méd Chile 2004; 132: 627-634
Los tumores neuroendocrinos del páncreas son de baja incidencia. En Norteamérica se diagnostican alrededor de 2.500 casos por año1. Se desarrollan desde las células de los islotes de Langerhans. Tanto los funcionantes como los no funcionantes son embriológica e histológicamente idénticos y sólo difieren en su capacidad para producir hormonas2.
Un número importante de ellos se malignizan, dan metástasis en el hígado y pueden incluso llevar a la muerte. Sin embargo, algunos como los no funcionantes malignos, tienen mejor pronóstico si se comparan con el adenocarcinoma pancreático3.
Un estudio comparativo del comportamiento de los tumores funcionantes y de los no funcionantes, no demuestra diferencia significativa en cuanto a incidencia de metástasis, índice de resecabilidad y tiempo de sobrevida libre de enfermedad4.
La ultrasonografía laparoscópica y la ultrasonografía intraoperatoria son de gran ayuda para localizar tumores pequeños no palpables5.
Actualmente, la cirugía es el único tratamiento curativo con sobrevida prolongada, siempre que se detecten tumores pequeños6. Si hay metástasis,se requieren otras terapias, dependientes de la rapidez de crecimiento tumoral y de la magnitud del trastorno hormonal. La quimioterapia actual no es muy exitosa7.
TUMORES FUNCIONANTES
Son secretores de uno o más péptidos activos. Hay una gran variedad de ellos: insulinoma, gastrinoma, glucagonoma, VIPoma (VIP= vasoactive intestinal peptide), somatostatinoma y ACTHoma (ACTH: Adrenocorticotropic hormone). Varios pueden ser tratados exitosamente mediante cirugía8. También hay tumores funcionantes asociados al síndrome de neoplasia endocrina múltiple (MEN-1) y a la enfermedad de von Hippel-Lindau.
Insulinoma. Tumor originado principalmente de las células pancreáticas en islote. Produce insulina en exceso. Tiene baja incidencia (4/1 millón de personas al año), pero es el tumor pancreático endocrino funcionante más frecuente (50 a 60%). Generalmente benigno de 2 cm de diámetro. Los malignos (5 a 10%) suelen ser mayores de 2,5 cm9. Son múltiples (10%), especialmente en pacientes con MEN 1. Habitualmente se presenta en la quinta y sexta década de la vida y más en mujeres que en hombres (2:1).
Cuadro clínico. Caracterizado por hipoglicemia del ayuno, secundaria a la continua secreción de insulina. Los síntomas neurológicos son los más característicos: visión borrosa, diplopía, amnesia, confusión, pérdida transitoria del conocimiento, coma en los casos más graves y daño neurológico permanente en hipoglicemias prolongadas. También hay signología por liberación de catecolaminas: ansiedad, temblor, sudoración, náuseas y palpitaciones. El diagnóstico puede ser tardío, por sintomatología mínima o por predominio de la signología neurológica10.
Diagnóstico. Actualmente, el diagnóstico se confirma por la asociación de hipoglicemia e hiperinsulinismo. Se realiza la prueba de ayuno controlado de 48 h o de 72 h: en ayuno total se mide la glicemia, insulinemia, péptido C y sulfonilurea. El ayuno termina, generalmente antes de las 48 h, cuando aparecen los signos de hipoglicemia. La prueba es positiva cuando la insulinemia es mayor de 6 mU/mL y la hipoglicemia es menor de 45 a 50 mg/dL11. Además está aumentado el péptido C (derivado de la proinsulina) sobre 0,2 nmol/L y no se detecta sulfonilurea. Es muy importante la disociación de los niveles plasmáticos de insulina y glucosa. No es posible afirmar la presencia de insulinoma si la hipoglicemia alcanzada por el ayuno no se acompaña de hiperinsulinemia. Es necesario descartar las múltiples causas de hipoglicemia9.
Para localizar el insulinoma se emplea ultrasonografía, tomografía, resonancia nuclear y endosonografía12. Durante la exploración quirúrgica se recurre a la palpación del páncreas, ultrasonografía intraoperatoria y pruebas especiales, como la determinación de insulina en sangre de venas hepáticas después de inyección intraarterial de calcio.
Los insulinomas no localizados por imagenología preoperatoria, entre 90 y 100% son detectados por exploración o ultrasonografía intraoperatorias13,14.
Tratamiento. Es esencialmente quirúrgico. Primero se ubica el insulinoma mediante exposición completa del páncreas, logrando así su visualización, palpación y exploración ultrasonográfica. La modalidad y magnitud de la resección dependen de la localización, tamaño y relación del tumor con estructuras vecinas como vena porta, vasos esplénicos o conducto pancreático. La intervención quirúrgica más utilizada es la enucleación del insulinoma.
La extirpación completa del tumor determina normalización de la glicemia e incluso tendencia a elevarse durante los primeros días. La persistencia de la hipoglicemia indica resección incompleta.
La experiencia nacional y extranjera muestran importante morbilidad. Veintisiete casos operados en la Universidad de Hong-Kong presentaron 33% de morbilidad y 0% de mortalidad en casos benignos15.
Cirugía con resección de mayor cantidad de tejido pancreático sólo se recomienda en las siguientes situaciones: tumores mayores de 2,5 cm (más malignos), compromiso del conducto pancreático y en insulinomas múltiples asociados al síndrome de MEN 1. Según la ubicación, se realiza pancreatoduodenectomía o pancreatectomía distal. Si en el preoperatorio no se localiza el tumor, no se justifica realizar pancreatectomía distal a ciegas porque habitualmente el insulinoma se detecta en el intraoperatorio16.
Autores holandeses17 y japoneses18 emplean ultrasonografía laparoscópica y enucleación por vía laparoscópica, para ubicar y tratar insulinomas.
Se recomienda resecar las metástasis hepáticas, si técnicamente es posible. Para las metástasis hepáticas irresecables, aconsejan quimioembolización arterial paliativa más resección del tumor primario19.
En Chile hay publicados 35 casos de insulinoma tratados mediante enucleación y resección pancreática. Los últimos 13 casos fueron publicados en los años 1997 y 2000. En ellos se realizaron 5 enucleaciones, 7 pancreatectomías distales y 1 pancreatoduodenectomía, con la siguiente morbilidad: 1 fístula biliar en el enfermo tratado mediante pancreatoduodenectomía, 2 fístulas pancreáticas de bajo débito y una fístula pancreática con colección intraabdominal que requirió reexploración quirúrgica. Todos los pacientes alcanzaron normoglicemia postoperatoria20-22.
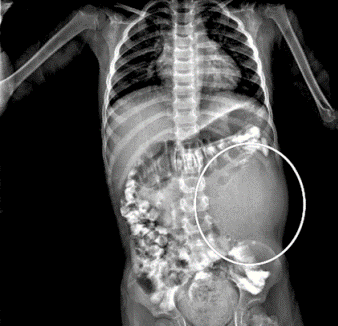
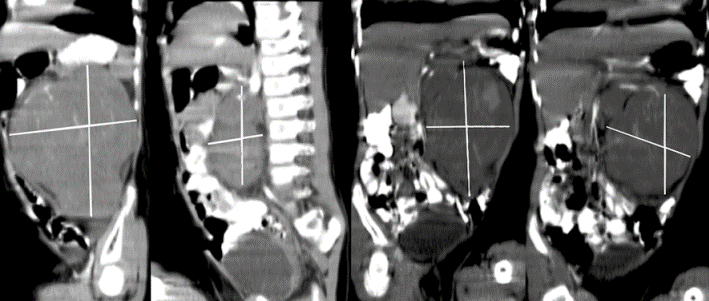
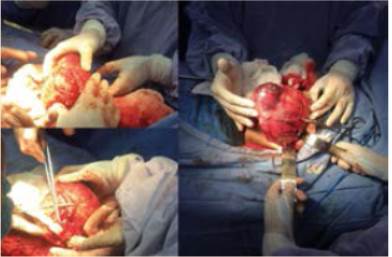
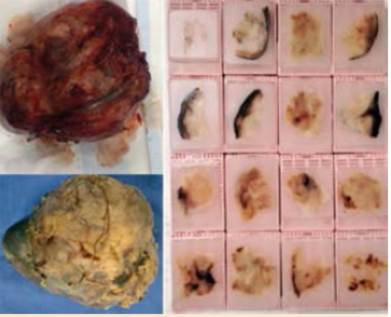
En el Servicio de Cirugía del Hospital Regional de Temuco entre 1990 y 1998 se operaron 4 pacientes con insulinoma benigno. Todos se diagnosticaron por el cuadro clínico y la prueba del ayuno de 72 h. Uno estaba en el proceso uncinado y los otros 3 en el cuerpo del páncreas. La ultrasonografía intraoperatoria ayudó a la ubicación exacta del tumor y descartó otra localización pancreática o metástasis hepáticas. Todos ellos fueron enucleados. Una paciente presentó una pancreatitis postoperatoria de curso grave, que mejoró después de realizar necrosectomía. Todos los pacientes normalizaron la glicemia e insulinemia. En la Figura 1 se visualiza un insulinoma de 1,8 cm del proceso uncinado, tratado por nosotros mediante enucleación.

Figura 1. Insulinoma de 1,8 cm ubicado en proceso uncinado del páncreas tratado mediante enucleación.
Gastrinoma. Es un tumor secretor de gastrina, al igual que el feocromocitoma y algunos tumores ováricos y broncogénicos. No obstante, sólo el gastrinoma es responsable de la gran hipergastrinemia del síndrome de Zollinger-Ellison9. De muy baja prevalencia (1 a 4 por 1 millón de habitantes), se presenta entre los 50 y 60 años, más en mujeres que en hombres (3:1). En 40% se localiza en el páncreas y en otro 40% en la pared duodenal. Se asocia al síndrome MEN 1 (20-25%) y entonces, frecuentemente, es maligno1.
Los gastrinomas primarios del hígado son raros, posiblemente serían localización secundaria de un gastrinoma pancreático o duodenal muy pequeño o involutivo23.
Cuadro clínico. El dolor abdominal es el síntoma más común y es secundario a úlcera péptica o reflujo gastroesofágico por hipergastrinemia. El 75% de estas úlceras se ubican en la primera porción duodenal. Otro síntoma es la diarrea, provocada por exceso de ácido que no alcanza a reabsorberse en el intestino delgado. Pueden también haber manifestaciones de hipergastrinemia acentuada: hemorragia digestiva, síndrome pilórico, perforación de úlcera péptica, hallazgo de úlceras en sitios inhabituales como el yeyuno proximal o ausencia de respuesta a los inhibidores de la secreción ácida. Tardíamente aparece caquexia y sintomatología propia del crecimiento tumoral.
Diagnóstico. Está dado por el cuadro clínico y los siguientes análisis:
1. Gastrinemia elevada más hipersecreción ácida en el ayuno.
2. Aumento de 200 pg/mL o más de gastrina sobre su nivel basal, posterior a la inyección de un bolo de secretina.
3. Prueba positiva de infusión cálcica: gastrina aumenta 395 pg/mL después de la infusión de calcio9. Es una buena prueba diagnóstica complementaria, especialmente cuando las otras son dudosas24.
La imagenología permite ubicar con gran exactitud gastrinomas menores de 1 cm de la pared duodenal. Primero se realiza cintigrafía de receptores de somatostatina para visualizar el tumor y posibles metástasis hepáticas y óseas. Se continúa con radiología contrastada del duodeno y endosonografía25. Una serie norteamericana demostró la gran efectividad de la endotes con síndrome de Zollinger-Ellison, se encontró que la úlcera péptica es el diagnóstico diferencial más frecuente (71%) y en 7%, respectivamente, el reflujo gastroesofágico y la diarrea idiopática crónica27. sonografía26.
En otra serie de 265 pacien
Tratamiento. Habitualmente se emplean inhibidores de la bomba de protones. El pantoprazol oral (40 a 160 mg/día) o intravenoso (160 a 240 mg/día) logran controlar el síndrome de Zollinger-Ellison28,29.
La cirugía es controversial. Algunos autores privilegian la terapia con inhibidores de la secreción ácida y otros la cirugía para evitar la diseminación tumoral que es la principal causa de mortalidad30. Cirujanos de la Universidad de California aconsejan resecar los gastrinomas de más de 2,5 cm, porque en un alto porcentaje desarrollan metástasis hepáticas31.
Los pacientes muy desnutridos por la enfermedad ulcerosa requieren alimentación paraenteral preoperatoria.
Durante la exploración quirúrgica se realiza exposición amplia del páncreas y duodeno para palpar el tumor con facilidad1. La ultrasonografía intraoperatoria ayuda bastante. La transiluminación duodenal intraoperatoria y la duodenectomía sirven para localizar los gastrinomas duodenales.
Los gastrinomas de la cabeza del páncreas pueden ser resecados mediante enucleación o pancreatoduodenectomía. La proximidad al conducto de Wirsung y la presencia de un tumor grande son indicación de pancreatoduodenectomía32.
En 4 a 7% de los pacientes no se logra ubicar el tumor. En ellos se indica vagotomía supraselectiva y en casos de enfermedad ulcerosa grave, excepcionalmente, se ha efectuado gastrectomía total.
Cuando aparecen metástasis hepáticas y si hay buenas condiciones generales, se indica hepatectomía para lograr mejor sobrevida a largo plazo33. También se han empleado exitosamente ondas de radiofrecuencia para destruir in situ las metástasis hepáticas34. En pacientes con gastrinoma metastásico diseminado, el uso de octreótido por largo tiempo ha demostrado ser más eficaz que el empleo de la quimioterapia tradicional35. También se han publicado casos tratados con trasplante hepático9.
Los gastrinomas asociados al síndrome MEN 1 preferentemente se tratan médicamente porque son habitualmente múltiples y malignos. La cirugía sólo se indica cuando el tumor tiene más de 3 cm1.
Somatostatinoma. Es un tumor generalmente maligno y extremadamente raro con sólo 200 casos publicados36. Es hipersecretor de somatostatina y en 10% secreta otras hormonas (gastrina, calcitonina, polipéptido intestinal vasoactivo, glucagón, insulina y ACTH). En 50% está en la cabeza del páncreas y, al momento de la cirugía, habitualmente tiene más de 5 cm, con metástasis hepáticas y ganglionares regionales. También se ubica en el duodeno, ampolla de Vater, conducto cístico y yeyuno.
Infrecuentemente aparece un síndrome caracterizado por diabetes mellitus, colelitiasis, esteatorrea e hipoclorhidria. La sintomatología es secundaria al crecimiento tumoral: dolor abdominal, baja de peso y trastornos digestivos poco definidos. Si la localización es extrapancreática, la clínica puede variar: ictericia si está en la ampolla de Vater o el conducto cístico y hemorragia digestiva u obstrucción intestinal si el tumor es de duodeno o yeyuno. Puede asociarse a la neurofibromatosis de von Recklinghausen y entonces, inicialmente se diagnostica feocromocitoma36.
Hay elevación de la somatostatina plasmática, pero habitualmente se diagnostica por exploración quirúrgica. La tomografía y resonancia magnética tienen una sensibilidad de más de 95% para los somatostatinomas pancreáticos mayores de 2 cm. También se utiliza la endosonografía y la cintigrafía con Indium111 para detectar receptores de somatostatina36.
La cirugía es la terapia de elección. La sobrevida a 5 años en ausencia de metástasis es de 100% y de 40% si hay metástasis ganglionares o hepáticas.
VIPoma. Tumor de baja incidencia, ubicado preferentemente en el páncreas. Es hipersecretor de polipéptido intestinal vasoactivo, el cual provoca el síndrome de Verner-Morrison o «cólera pancreática»: diarrea hipersecretora, rubicundez, hipokalemia y aclorhidria9.
Se diagnostica cuando hay un tumor pancreático de varios centímetros, presencia de diarrea acuosa profusa (2 a 3 litros/día) y elevación del polipéptido vasoactivo intestinal9.
El tratamiento comienza con reposición hidroelectrolítica y bloqueo de la hiperfunción hormonal mediante octreótido. La terapia definitiva es la cirugía y la modalidad de resección depende de la localización del tumor. Recientemente, se realiza enucleaciones y pancreatectomías distales laparoscópicas, en pacientes portadores de diferentes tipos de tumores neuroendocrinos incluidos los VIPomas37.
Glucagonoma. Tumor generalmente maligno que produce aumento del glucagón secretado por las células alfa en islotes. De incidencia muy baja, con alrededor de 200 casos publicados38.
Entre los cincuenta y setenta años se manifiesta con diabetes mellitus, tromboembolismo y eritema necrolítico migratorio muy pruriginoso. Además hay glositis y trastornos neuropsiquiátricos como ataxia, hiperreflexia, depresión, psicosis, demencia e incontinencia esfinteriana. El tumor puede dar metástasis ganglionares, hepáticas, óseas, suprarrenales y pulmonares38.
El diagnóstico se sospecha por la clínica y se confirma por el valor elevado de glucagón plasmático (>1.000 pg/mL). También puede detectarse hipoalbuminemia, hipoaminoacidemia, anemia e hipocolesterolemia. La tomografía visualiza tumores de varios centímetros ubicados especialmente en el cuerpo del páncreas.
El tratamiento ideal es la cirugía. Según su tamaño se efectúa enucleación, pancreatectomía distal con preservación esplénica o pancreatoduodenectomía. El octreótido u otros análogos de la somatostatina sirven para frenar la hipersecreción hormonal. El tromboembolismo pulmonar se previene con implante de un filtro en la vena cava inferior. En pacientes con metástasis hepáticas, a pesar de ser controversial, se ha realizado trasplante hepático más resección del tumor primario39.
ACTHoma. Los tumores de células en islotes secretores de ACTH sólo se encuentran en el páncreas. Son de crecimiento rápido y precozmente presentan metástasis. Se produce una forma muy virulenta de síndrome de Cushing y por la producción agregada de gastrina, pueden desarrollar síndrome de Zollinger-Ellison. En general son de mal pronóstico, porque no responden a la terapia sistémica y el empleo de octreótido más ketoconazol es sólo paliativo9.
Neoplasia endocrina múltiple tipo 1. Este síndrome de baja prevalencia (0,02 a 0,2 por 10.000), es provocado por un trastorno genético familiar autosómico dominante, con mutación de un gen oncosupresor localizado en el cromosoma 11q. Hay hiperplasia o adenoma de las paratiroides (90%), tumores endocrinos pancreatoduodenales (30 a 80%) y en alrededor de 33%, adenoma de la pituitaria, tumores adrenocorticales secretores o tumores no funcionantes. Se presenta entre los 20 a 40 años de edad, con igual frecuencia en ambos sexos9.
Clínicamente, la enfermedad se manifiesta por: síndrome de Zollinger-Ellison secundario a la hipergastrinemia, hipoglicemia determinada por el insulinoma y otros síntomas dados por el crecimiento local de tumores no funcionantes.
El hiperparatiroidismo, generalmente, da sus manifestaciones entre los 30 y 40 años de edad: nefrolitiasis, osteoartralgias, dolores abdominales, fatiga muscular y trastornos neuropsiquiátricos. Los niveles de calcio y hormona paratiroídea están elevados.
La mejor prueba para diagnosticar tumores endocrinos del páncreas en pacientes con sospecha de MEN 1, es la medición de polipéptido pancreático y gastrina al doble de sus valores normales después de un desayuno abundante en carbohidratos y reducido en proteínas9. Para visualizar el tumor se emplea la tomografía helicoidal, endosonografía, cintigrafía de receptores de somatostatina y resonancia magnética. La funcionalidad del o los tumores se demuestra con la determinación de insulina en ayuno de 48 a 72 h, test de secretina y de atropina.
El tratamiento busca el control de la hipersecreción ácida gástrica mediante inhibidores de la secreción ácida. En algunos centros recomiendan la cirugía cuando los tumores alcanzan 3 cm de diámetro por el mayor riesgo de malignidad. Según la ubicación y el tamaño del tumor hay varias opciones: enucleación, resección de la submucosa duodenal si los tumores se ubican preferentemente en el duodeno, pancreatectomía parcial o pancreatoduodenectomía. Sin embargo, ya puede haber desarrollo de metástasis, como se mostró en una serie de 21 pacientes operados con 33% de metástasis linfáticas regionales40. Por este motivo, algunos autores recomiendan una conducta quirúrgica más agresiva.
La paratiroidectomía se indica cuando aparece el hiperparatiroidismo, con lo cual disminuye la calcemia y como ésta estimula la producción de gastrina, en muchas oportunidades la paratiroidectomía mejora el Zollinger-Ellison o al menos determina menor consumo de inhibidores de la secreción ácida9.
TUMORES NO FUNCIONANTES
Representan entre 35 y 50% de los tumores neuroendocrinos del páncreas, se diagnostican entre la quinta y sexta década de la vida y tienen igual distribución por sexo. En el 50% se ubican en la cabeza, proceso uncinado y cuello del páncreas41. Son de crecimiento lento y detectados cuando alcanzan gran tamaño, con infiltración de órganos vecinos más desarrollo de metástasis hepáticas y ganglionares regionales. Tienen producción hormonal muy escasa o son no secretores9.
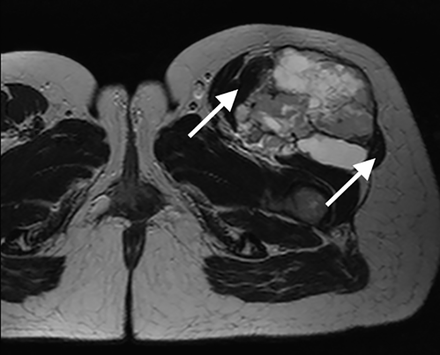
Los pacientes presentan pérdida de peso, dolor abdominal, masa palpable e ictericia. A veces hay complicaciones como hemorragia digestiva masiva por erosión del tubo digestivo o vasos retroperitoneales. Los tumores son visualizados por tomografía helicoidal y resonancia magnética. Es importante diferenciarlos del adenocarcinoma ductal y otros tumores mucinoso quísticos del páncreas9. El Ppoma (productor de polipéptido pancreático) es el tumor no funcionante más frecuente.
La cirugía es el tratamiento de elección. Por el gran tamaño del tumor se indica resección extensa, más linfadenectomía regional, para disminuir la recurrencia tumoral. Las metástasis hepáticas bien delimitadas deben ser resecadas, incluso, se han realizado trasplantes hepáticos. En pacientes con tumor irresecable se recomienda efectuar derivación gastrointestinal o biliodigestiva. La quimioterapia paliativa (estreptocina y doxorrubicina) muestra buenos resultados9. La quimioembolización es otra alternativa para tratar pacientes con metástasis hepáticas irresecables.
TUMORES PANCREÁTICOS FAMILIARES
Enfermedad de von Hippel-Lindau. Es un síndrome neoplásico hereditario causado por mutación de un gen oncosupresor ubicado en la rama corta del cromosoma 3. La incidencia es de 1 en 31.000 a 36.000 nacidos vivos. Existe alto riesgo de desarrollar tumores vasculares del sistema nervioso central y de la retina, carcinoma renal de células ciaras, tumores neuroendocrinos del páncreas, feocromocitoma, tumores saculares endolinfáticos y quistes benignos de variadas localizaciones. Son frecuentes los quistes pancreáticos múltiples, habitualmente asintomáticos, salvo casos aislados con obstrucción duodenal o insuficiencia pancreática.
En familias con historia de von Hippel-Lindau, los tumores se detectan con ultrasonografía o tomografía una vez al año desde los 11 años de edad y tomografía anual después de los 20 años de edad. Idealmente, el diagnóstico debe ser precoz para llegar con la cirugía resectiva antes de la aparición de metástasis. Igualmente deben buscarse otras manifestaciones de la enfermedad. El análisis genético también está dirigido al diagnóstico precoz de los diversos tumores de esta enfermedad.
Los tumores neuroendocrinos pancreáticos generalmente son no secretores y tienen indicación de cirugía resectiva por riesgo elevado de malignidad42,43.
CONCLUSIÓN
Aun cuando los diferentes tumores neuroendocrinos del páncreas tienen identidad propia, es recomendable seguir una pauta general en su diagnóstico y tratamiento.
1. Primero es necesario precisar cuáles son las alteraciones bioquímicas responsables del síndrome endocrino. Si sospechamos un insulinoma, es mejor iniciar el diagnóstico mediante la prueba de ayuno de 48 ó 72 h y a continuación realizar los procedimientos de localización. No comenzar la búsqueda con una laparotomía exploradora, porque los síntomas de hipoglicemia y tomografía sospechosa de insulinoma, podrían corresponder a otras patologías.
2. Es importante determinar si hay antecedentes familiares de enfermedad porque entonces es frecuente encontrar patología endocrina múltiple, localizaciones fuera del área pancreatoduodenal y malignidad.
3. Según el tipo de tumor, se deben tratar los síndromes hormonales antes de ir a una terapia más definitiva. Así por ejemplo, el tratamiento del síndrome de Zollinger-Ellson con inhibidores de la secreción ácida es actualmente bastante efectivo.
4. La imagenología actual permite localizar tumores pancreáticos neuroendocrinos muy pequeños. En Chile es posible recurrir a tomografía axial computada helicoidal, resonancia nuclear magnética, cintigrafía, endosonografía y ultrasonografía intraoperatoria. Los procedimientos más invasivos como la angiografía, son menos usados.
5. Es necesario bloquear los efectos de la disfunción endocrina en el preoperatorio de los pacientes que van a ser intervenidos, especialmente en los insulinomas y gastrinomas.
6. Aún es controversial indicar cirugía resectiva por potencial malignidad cuando la disfunción endocrina puede mejorar con terapia médica, como ocurre en el gastrinoma.
7. En el síndrome de MEN 1, la indicación y tipo de cirugía a emplear es motivo de controversia. Se determina por la edad, estado del paciente, magnitud del cuadro endocrinológico, riesgo de metástasis o presencia de diseminación durante la exploración quirúrgica. Recientemente, se descubrió el gen del síndrome de MEN 1. Esto permitirá en el futuro ir a la demostración temprana de tumores pequeños y en consecuencia, indicar cirugía más conservadora para preservar la función exocrina y endocrina, especialmente del páncreas44.










{kind=link}